
Adding bioinformatic software to Bioconda - a reference guide
In this first part of a three part guide, this post aims to provide an (opinionated) guide to adding a tool or package to Bioconda.
- For part two of this guide, see updating a package on Bioconda.
- For part three of this guide, see debugging a Bioconda build.
The conda package manager combined with the Bioconda repository has become a de facto gold-standard way for distributing bioinformatics software (Grüning et al. 2018). The associated Biocontainer project serves to provide complementary Docker and Singularity containers from the same conda (da Veiga Leprevost et al. 2017).
While the Bioconda team has provided a huge amount of impressive infrastructure to make adding our bioinformatic tools and packages to the repository as easy as possible, the documentation is lacking at points. In particular, while the Bioconda documentation explains nicely how to contribute to the repository, I personally found that it misses out on the important part of how to make what we actually will add i.e, how to make the conda recipe specifically for Bioconda 1. Another thing I found particularly tricky is how to debug the build process if things go wrong.
I hope to here provide guidance, based on my personal experience, to people who are interested in adding their software to Bioconda but maybe felt it too overwhelming to start. Hopefully it will provide sufficient ‘hand-holding’ to get us through the process, after which it’s just ‘rinse and repeat’!
The main sections of this post are:
- TL;DR
- Prerequisites
- Adding a new tool or package
- Debugging a recipe build
- Updating an existing tool or package recipe
Please note that this post comes with ‘no warranty’!(!), as the Bioconda build steps could change at any point. However, the steps here should act as a good starting point. Furthermore, if you are planning to add someone else’s tool or package to Bioconda, it’s always good etiquette to ask or just inform the original authors that it will happen.
I would like to thank George Bouras () for prompting the formalisation of my rough notes that he shared on Twitter where it became more popular than I expected.
TL;DR
Overview
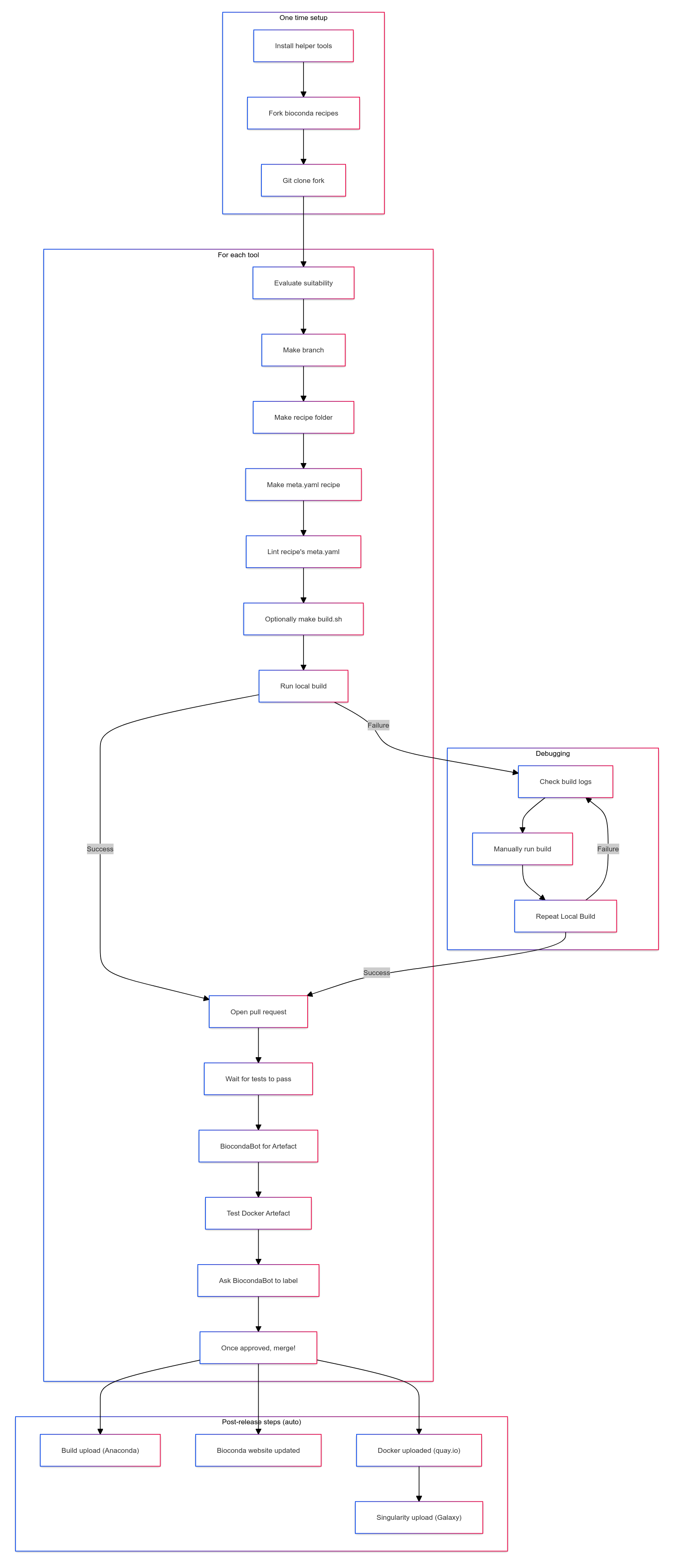
Relevant Commands
## Create environment with conda building tools
conda create -n bioconda-build -c conda-forge -c bioconda conda-build bioconda-utils greyskull
conda activate bioconda-build
## Clone repo of your fork of https://github.com/bioconda/bioconda-recipes and make branch
git clone <your-forked-bioconda-recipes-repo-address>
git switch -c add-<toolname>
## Make recipe meta.yaml
## Option 1: If using Greyskull
cd recipes/
greyskull <pypi/cran> <toolname>
## Option 2: If not using Greyskull
mkdir recipes/<toolname>
touch recipes/<toolname>/meta.yaml
## Lint recipe meta.yaml
bioconda-utils lint recipes/ --packages pathphynder
## Perform a local build (two options)
## Option 1:
conda build recipes/<toolname>
## Option 2:
bioconda-utils build --docker --mulled-test --packages <toolname>
## Debugging
## Option 1: conda-build
cd /<path>/<to>/<conda-install>/envs/<toolname>/conda-bld/linux-64
conda create -n <debugging-env-name> -c ./ <tool_name_as_in_recipe>
conda build recipes/<toolname> --keep-old-work
## Option 2: bioconda-utils
docker run -t --net host --rm -v /tmp/tmp<randomletters>/build_script.bash:/opt/build_script.bash -v /<path>/<to>/<conda-install>/envs/<toolname>/conda-bld/:/opt/host-conda-bld -v /<path>/<to>/<recipes_local_clone>/recipes/<toolname>:/opt/recipe -e LC_ADDRESS=en_GB.UTF-8 -e LC_NAME=en_GB.UTF-8 -e LC_MONETARY=en_GB.UTF-8 -e LC_PAPER=en_GB.UTF-8 -e LANG=en_GB.UTF-8 -e LC_IDENTIFICATION=en_GB.UTF-8 -e LC_TELEPHONE=en_GB.UTF-8 -e LC_MEASUREMENT=en_GB.UTF-8 -e LC_TIME=en_GB.UTF-8 -e LC_NUMERIC=en_GB.UTF-8 -e HOST_USER_ID=1000 quay.io/bioconda/bioconda-utils-build-env-cos7:2.11.1 bash
conda mambabuild -c file:///opt/host-conda-bld --override-channels --no-anaconda-upload -c conda-forge -c bioconda -c defaults -e /opt/host-conda-bld/conda_build_config_0_-e_conda_build_config.yaml -e /opt/host-conda-bld/conda_build_config_1_-e_bioconda_utils-conda_build_config.yaml /opt/recipe/meta.yaml 2>&1
conda activate /opt/conda/conda-bld/<toolname_hash>/_build_env
## Testing the Docker image artifact
docker run -it <image_id_from_docker_images_command>
Prerequisites
-
Make a fork of the bioconda-recipes GitHub repository, and clone this to our local machine 2.
-
Install on our local machine the following software:
condaitself- Bioconda configured as a source channel (see bioconda documentation)
-
The following conda packages:
conda-utilsbioconda-buildgreyskull(optional: for Python software on pypi or R packages on CRAN)
I typically dump all of the above in a specific conda environment, generated with the following command:
conda create -n bioconda-build -c conda-forge -c bioconda conda-build bioconda-utils greyskull conda activate bioconda-build docker(optional: for local build testing)
Adding a new tool or package to Bioconda
Preparation
-
Ask: is my software already on Bioconda?
- Search the Bioconda website https://bioconda.github.io/ to make sure some kind soul hasn’t already done this.
- Also double check the software doesn’t already exist on another conda channel on Anaconda.
-
Ask: Is the software right for Bioconda?
- Bioconda is for bioinformatics software.
- If the tool is a more generic tool or for a different domain, we may want to consider adding it to conda-forge 4.
- One common caveat to this is R packages - if our biology-related package is on CRAN (https://cran.r-project.org/), it should go on conda-forge, if it’s on Bioconductor (https://www.bioconductor.org/) it should go on Bioconda (if it’s not already there).
-
Check: Does the software have a compatible license? (i.e., allows redistribution)
-
Check: Does the software have a stable release?
- I.e., an unmodifiable file (tarball or zip) and stable URL that that specific version can be always be downloaded from.
- An example is a GitHub release (e.g. for a Kraken2 release, we use the link of the ‘Source code (tar.gz)’, i.e.,: https://github.com/DerrickWood/kraken2/archive/refs/tags/v2.1.3.tar.gz).
- Using GitHub ‘tags’ are sort of OK.
- Using specific commits (i.e., no versioned release tarballs) are strongly frowned upon.
If we are all good with the above, we can put our tool or package on Bioconda.
Writing the recipe
A Bioconda recipe at a minimum can consist of a single file called meta.yaml.
This is often sufficient for PyPi Python and many R packages (respectively).
-
Create a new git branch for the tool we wish to add within the forked and cloned
bioconda-recipesrepository:git switch -c add-<toolname> -
Make a
meta.yamlfile within the created directory, with one of two methods:-
If the tool is a Python package on pypi or a R package on CRAN, we can use
greyskullto generate this for us.cd recipes/ greyskull <pypi/cran> <toolname> -
In all other cases, make a new directory in the
recipes/directory, named after the software we wish to add.mkdir recipes/<toolname>The name of the software must be formatted in all lower case, and with only letters, numbers, and hyphens.
If our package is an R package, we should prefix the name with
r-.⚠ Make sure a tool with the same name doesn’t exist! If it does - consider adding a suffix. For example,
-mgto indicate software for metagenomics, or-litefor a version of a recipe that doesn’t include preinstalled databases.Then, create an empty text file called
meta.yamlin the new directory.touch recipes/<toolname>/meta.yaml
-
-
Add the following sections in the
meta.yamlfile (or double check if already made withgreyskull). When in doubt, copy from other similar existing recipes already on Bioconda:package:- Specify the name (same specifications as above) and version of the tool/package.
source:- Specify the URL to the source code tarball or zip file for conda to download.
- The e.g.
sha265hash string of the file for download verification.
build:- Specify the build number (for new packages or new software version, always
0). - Possibly the architecture (e.g.
noarchfor Python packages). - A
run_exportssubpackage pinning.
- Specify the build number (for new packages or new software version, always
requirements:- Specify a list of the various dependencies of the software needs during various sections of the build process, i.e.,
host,build, andrun. - Should have a minimum versions, and ideally a with
>=notation.
- Specify a list of the various dependencies of the software needs during various sections of the build process, i.e.,
test:- One or more (e.g. if multiple CLI tools or scripts exist under the package) commands to test the software installed correctly.
- Typically simply running the tool with
--helpor--versionis sufficient, but must have a0exit code to indicate success. - If
--helpends with a non-0code, we can trygreping for a string in the help message.
about:- URL of such as source code repository or documentation home page.
- License type 5.
- Corresponding license file name as in the tarball.
- A short one-sentence summary and/or long-form description of the software.
extras:- other metadata information such as the DOI identifier of any associated publication the software may have.
- Other identifiers of the software.
An example of a
meta.yamlis as follows:{% set name = "centrifuge" %} {% set version = "1.0.4.1" %} package: name: {{ name|lower }} version: {{ version }} build: number: 2 skip: true # [osx] run_exports: - {{ pin_subpackage("centrifuge", max_pin="x.x") }} source: url: https://github.com/DaehwanKimLab/centrifuge/archive/refs/tags/v{{ version }}.tar.gz sha256: 638cc6701688bfdf81173d65fa95332139e11b215b2d25c030f8ae873c34e5cc patches: - centrifuge-linux-aarch64.patch # [linux and aarch64] requirements: build: - make - {{ compiler('cxx') }} host: - zlib run: - zlib - perl - wget - tar - python test: commands: - centrifuge --help about: home: https://github.com/DaehwanKimLab/centrifuge license: GPL-3.0-only license_file: LICENSE license_family: GPL3 summary: 'Classifier for metagenomic sequences. Supports classifier scripts' extra: additional-platforms: - linux-aarch64 identifiers: - biotools:Centrifuge - doi:10.1101/gr.210641.116A relatively simple example conda recipe example for Centrifuge, based on the descriptions above.
-
Lint our
meta.yamlfor any errors pertaining to Bioconda linting guidelines (make sure we’re in the root of the repository!).bioconda-utils lint recipes/ --packages <toolname>If there are any errors, I recommend fixing them before proceeding, as getting the same errors during the Bioconda GitHub CI takes a long time (as we’ll see later). In particular, the
missing_run_exportsis a new linting check that has been added recently, that many people are not aware of. To solve this one, look at recently merged recipes, as the PR template describes how to set this under ‘Instructions for avoiding API, ABI, and CLI breakage issues’, such as on thispango-collapsePR.
Writing a build script (optional)
For some tools, we may also need to create a build.sh script 6 in the same directory alongside the meta.yaml file.
This is simply a shell script that is run during the build process after cloning of the source code. The commands executed in this script are run in a specific build environment.
The purpose of this script varies, so I can’t give a precise definition or explicit steps for writing one, but in my experience it is most often used in cases of:
- Tools that need to be compiled from source code (e.g. C++ tools and
make install). - Tools that are simply just an executable binary that needs to be linked or copied to the
bin/of the eventual conda environment (e.g. Java.jarfiles). - Tools that have additional ‘auxiliary’ or ‘helper’ scripts outside of (and in addition to) the main tool that also need to be copied to the
bin/of the eventual conda environment. -
Patching files to allow them to run (often for simple patching with e.g.
sed, more complex patching can use a git stylepatchfile specified in themeta.yaml).- Patching can be stuff like adding a
shebangat the top of a file - Replacing hardcode paths or variables in
makefiles etc.
- Patching can be stuff like adding a
- Tools that may require other files to be copied to other directories in the conda environment (e.g. databases).
You can see an example of a build.sh script below:
#!/bin/bash
set -xe
export LDFLAGS="-L$PREFIX/lib"
export CPATH=${PREFIX}/include
mkdir -p $PREFIX/bin
case $(uname -m) in
aarch64)
CXXFLAGS="${CXXFLAGS} -fsigned-char"
ARCH_OPTS="SSE_FLAG= POPCNT_CAPABILITY=0"
;;
*)
ARCH_OPTS=""
;;
esac
make -j ${CPU_COUNT} CXX=$CXX RELEASE_FLAGS="$CXXFLAGS" ${ARCH_OPTS}
make install prefix=$PREFIX
cp evaluation/{centrifuge_evaluate.py,centrifuge_simulate_reads.py} $PREFIX/bin
A relatively simple example build.sh script for Centrifuge, based on the descriptions above. Here it includes both make install compilation examples with Bioconda C++ environment variables and copying of the additional auxiliary scripts to the bin/ directory.
However, as always, check other tools/packages for examples.
Examples of small build.sh scripts from the four examples above:
- kallisto (make install).
- MALT (java jar file).
- metabinner (auxiliary scripts).
- phynder (patching).
- grid (database files).
To provide further guidance based on my experience:
The $PREFIX variable corresponds to the the root of the conda environment that eventually gets made on a users system when they install the conda package.
You can explore our own conda environments to see what the $PREFIX looks like by running conda env list to see all of our own conda environments, and changing into the one of the directory listed in there.
They often will look very similar to Unix root directories, with folders such as etc/, bin/, lib/, share/, etc.
for example, if we have an executable or scripts that need to go into bin/, we must copy this into $PREFIX/bin.
For some tools we may have to copy other files into other directories, such as databases 7, but this is less common.
Another tricky thing is compiling of C++ code, which can be a bit of a pain.
For reasons 8, we need to use specific variables that point to the non-standard (it seems) places that conda stores its libraries and headers.
These are described here, and in particular for zlib.
You often will need to patch the make files and other compilation related scripts to use these variables, and also to use the --prefix=$PREFIX flag when running make install.
For all of the above, regardless of language, I recommend looking at the the contributor guidelines.
Build testing
Once we think we’ve got our meta.yaml and build.sh (if needed) files ready, we can now try to see if this works.
We have two options here, either:
- Test it locally (less slow, but may not perfectly replicate the build).
- Open the pull request onto the main
bioconda-recipesrepository and see if it passes the tests there (slow).
If we want to just let the Bioconda CI do the testing, skip to the next section.
Otherwise, in our Bioconda-build conda environment, we can run one of two options (in both cases from the root directory of our `bioconda-recipes fork):
-
The standard
conda buildcommand:conda build recipes/<toolname> -
The
bioconda-utilscommand, which should better replicate the CI environment and also gives us the Biocontainer Docker version of our conda environment (but requires Docker, and is slower):bioconda-utils build --docker --mulled-test --packages <toolname>
Hopefully, if everything worked correctly the first time, we should have a successful build and we can proceed with submitting to bioconda. If something goes wrong, see part 3 of this guide on debugging the Bioconda builds.
Regardless, in both local build approaches, these commands will dump a huge amount of output to the terminal, and if it fails, we’ll have to trawl through it to debug it.
I generally find the bioconda-utils method is slightly easier to debug because of the use of colours in the logging, with added benefit of making it easier to check the Biocontainer Docker image that gets created, but which method is up to personal preference.
Opening the Pull Request
Once we’re happy with our recipe, we can open a pull request on the main bioconda-recipes repository on GitHub.
We can do this (if you’re not too familiar with GitHub), by:
- On your local repo,
git adding the files you’ve added, commit, and push. - Go to the main
bioconda-recipesrepository on GitHub. - Switch to the Pull Requests tab.
- Press the green ‘New Pull Request’ button.
- In the top bar use the dropdowns to select our fork and branch (which should then be going into
bioconda/bioconda-recipesand themasterbranch). - Make sure the title of the pull request is follows the recommendations, typically just
Add <tool/package>orUpdate <tool/package>. - Once we open the pull request, the Bioconda CI will run.
We can see the overall status of the checks near the bottom of the page below the ‘Review required’ message. For most builds this currently happens away from GitHub on Microsoft Azure, and can take a while (sometimes up to 1 hour!) to complete (so be patient).
To get more information on the status of the CI test, and also logs, press ‘details’ next to one of the checks (it generally doesn’t matter which one), then press the ‘View more details on Azure Pipelines’ link on the resulting page.
On the Azure website we should see a series of ‘stages’, that run in order. The tests that are run in these stages are:
lint: checks we’ve not missed anything (e.g. the LICENSE).test_linux: that the recipe builds on a Linux system (i.e., doesn’t error and the test command completes).test_osx: that the recipe builds on a macOS system (i.e., doesn’t error and the test command completes).
A given stage has a completed (green tick), running (blue spinny icon), or failed (red cross) status. If we click on any of the stages, we should see log files that similar or identical what we would do if we were building locally (see that section for debugging advice, if we skipped local building).
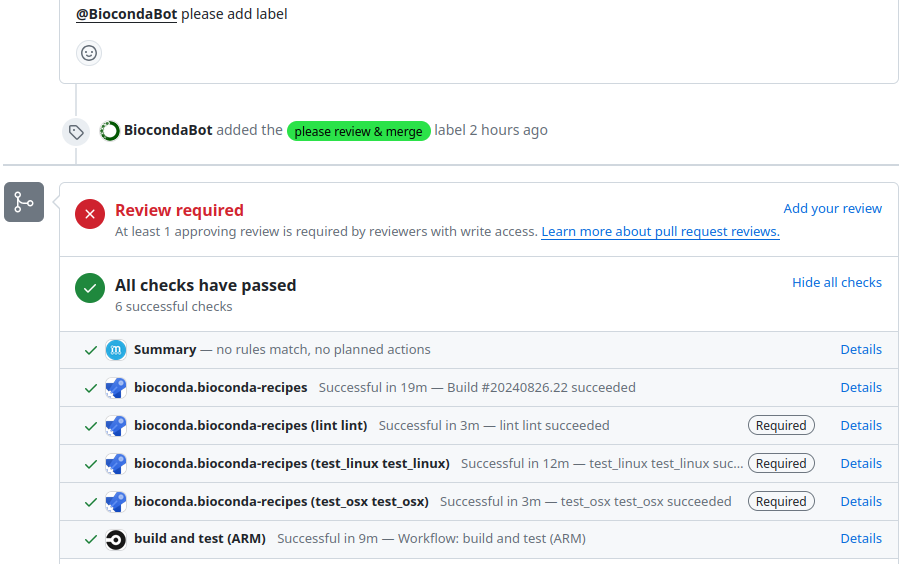
Screenshot of bottom of a GitHub PR with the checks list displayed with blue ‘Details’ links next to each test
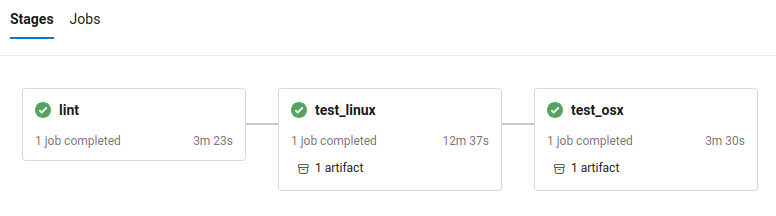
Screenshot of the Microsoft Azure interface with the three (successful) Bioconda CI stages.
If you get errors or something goes wrong, see part 3 of this guide on how to locally debug the Bioconda build.
Test driving the docker Biocontainer (optional)
If we used the bioconda-utils command to build our recipe, we can also optionally test the Biocontainer Docker image that was generated from the conda environment that was built.
If we did a local build, the Docker image is already on our own machine.
If we let the automated Bioconda CI do the testing on Azure, we can leave a comment with ‘ please fetch artifacts’ and this will generate a comment on the PR with two tables.
We can download the
LinuxArtifacts.zip file from the top table (Package(s) built are ready...), unzip it and then run the command given in Docker image(s) built table to load the container.
Then for both local or GitHub build cases, we can just access the created Docker container by finding it in the the output of docker images.
The image will be named something like quay.io/biocontainers/<toolname>, and I typically run the following command to access container and run additional test commands or experiments within the container.
docker run -it <image_id_from_docker_images_command>
This should dump us within a shell in the container so we can test commands etc. as we would with any other Docker container.
If something goes wrong here and you encounter issues with the build within the container, you can see part 3 of this guide to get tips and tricks how to manually re-build the recipe step-by-step. Otherwise, if you’re happy you can continue to finalise the PR in the next section.
Finalising the PR
If the CI on Microsoft Azure passes, then back on GitHub we can leave a comment in our PR saying ‘ please add label’.
This will add a label to our PR indicating a Bioconda team member can review our recipe to ensure it matches the guidelines.
If they give an approval, they or we can merge our PR into the main
bioconda-recipes repository!
We’re now officially a Bioconda recipe maintainer 🎉.
Once the recipe is merged in, we can normally install the official version of our tool/package with conda within a few minutes.
At the same time, on merging, the auto-generated Docker Biocontainer gets uploaded to the Biocontainers quay.io repository.
For the Singularity version of the Docker container, this can take up to 24h before it’s visible on the Galaxy project’s ‘depot’.
Conclusion
This part one of this guide hopefully has given you enough pointers on the steps required to make a recipe and submit your tool/package to Bioconda.
In the second part of this guide, we will go through how to update an existing recipe. In the third part, we will go through how to manually debug the build process if things go wrong.
As with all bioinformatics and software development in general, things rarely just ‘work’ straight out of the box. My three biggest points of advice:
- Always copy and paste from other similar tools or packages on the Bioconda recipes repository.
- Take the time to read through the whole log messages (sometimes you can find critical clues hidden amongst the verbose information).
- Take the time to go step by step trying to follow exactly what Bioconda does during it’s own building on Azure with local building.
I found by taking the time, I very quickly learnt common issues and how to solve them. However, if you’re really stuck (even after reading the third part of this guide), you can always ask the very friendly volunteer Bioconda team on the Bioconda gitter/matrix channel.
Footnotes
-
conda-forgeis another very useful and popular conda channel, but it is more general and not specifically for bioinformatics software, and has a different process for adding tools ↩ -
You can do a shallow clone
git clone --depth 1, to make the size of the cloned repo smaller on your machine. Thanks tofor the tip! ↩
-
Various Bioconda documentation pages say we should use
mamba, but recent versions of conda includelib-mambaby default, so generally we can use standardconda. But if you’re having problems with things being very slow, try switching tomamba. ↩ -
Note that conda-forge has a different system for adding packages! ↩
-
Possibly from a fixed list, and how to format these, I don’t know… I just copy and paste from other recipes. ↩
-
I’ve noticed in a few more recent recipes that these commands can go within the
meta.yamlitself in an entry calledscript:underbuild:, but I guess this only works for very simple commands… ↩ -
Even though I absolutely HATE this, as often it leads to gigantic multi-gigabyte conda environments which we can’t use on small CI runners. Give me the choice where to store my databases already! Don’t force me to place them in a specific place /rant. ↩
-
That I’ve never found a good explanation or documentation for. ↩
References
- Veiga Leprevost, F. da, B. A. Grüning, S. Alves Aflitos, H. L. Röst, J. Uszkoreit et al., 2017 BioContainers: an open-source and community-driven framework for software standardization. Bioinformatics (Oxford, England) 33: 2580–2582. 10.1093/bioinformatics/btx192 https://academic.oup.com/bioinformatics/article-pdf/33/16/2580/49041124/bioinformatics_33_16_2580.pdf
- Grüning, B., R. Dale, A. Sjödin, B. A. Chapman, J. Rowe et al., 2018 Bioconda: sustainable and comprehensive software distribution for the life sciences. Nature Methods 15: 475–476. 10.1038/s41592-018-0046-7
Citing this Post
@misc{adding-to-bioconda-quickguide,
author = "",
title = "Adding bioinformatic software to Bioconda - a reference guide",
year = "2024",
month = "08",
day = "16",
url = "https://ubinfie.github.io/2024/08/16/adding-to-bioconda-quickguide.html",
note = "[Online; accessed TODAY]"
}
Comments